






lundi, 14 décembre 2020
Percée majeure dans notre compréhension du repliement des protéines
 Le 30 novembre dernier, les résultats d’une compétition de prédiction de la structure des protéines appelée « CASP » (Critical assessment of structure prediction) ont été rendus publics. À chaque deux ans, une centaine d’équipe dans le monde tentent de découvrir à l’aide de logiciels la structure tridimensionnelle de certaines protéines que l’on a réussi à identifier par d’autres moyens expérimentaux. Depuis 2006, les taux de succès de ces logiciels oscillaient entre 30 et 40%. Puis, en 2018, le logiciel AlphaFold de la branche Deep Mind de Google, qui utilisent les réseaux de neurones artificiels (le fameux « deep learning »), a fait une entrée fracassante dans la partie avec un taux de succès de près de 60% ! C’est la même approche que Deep Mind avait déployé dans son logiciel AlphGo qui avait battu en 2016 le champion du monde du jeu de Go. Or les performances d’AlphaFold(2), la version 2020 du logiciel, lors de la compétition de cette année marquent, selon certains, un tournant majeur dans l’un des plus grands défis de la biologie puisque le logiciel a décrit la structure tridimensionnelle des protéines avec un taux de succès médian de 92% pour l’ensemble des protéines soumises et de 87% pour les protéines dont la structure était jugée comme étant particulièrement difficile à résoudre ! Est-ce le début d’une nouvelle ère ? C’est ce qu’affirment en tout cas certaines personnes qui travaillent dans le domaine.
Le 30 novembre dernier, les résultats d’une compétition de prédiction de la structure des protéines appelée « CASP » (Critical assessment of structure prediction) ont été rendus publics. À chaque deux ans, une centaine d’équipe dans le monde tentent de découvrir à l’aide de logiciels la structure tridimensionnelle de certaines protéines que l’on a réussi à identifier par d’autres moyens expérimentaux. Depuis 2006, les taux de succès de ces logiciels oscillaient entre 30 et 40%. Puis, en 2018, le logiciel AlphaFold de la branche Deep Mind de Google, qui utilisent les réseaux de neurones artificiels (le fameux « deep learning »), a fait une entrée fracassante dans la partie avec un taux de succès de près de 60% ! C’est la même approche que Deep Mind avait déployé dans son logiciel AlphGo qui avait battu en 2016 le champion du monde du jeu de Go. Or les performances d’AlphaFold(2), la version 2020 du logiciel, lors de la compétition de cette année marquent, selon certains, un tournant majeur dans l’un des plus grands défis de la biologie puisque le logiciel a décrit la structure tridimensionnelle des protéines avec un taux de succès médian de 92% pour l’ensemble des protéines soumises et de 87% pour les protéines dont la structure était jugée comme étant particulièrement difficile à résoudre ! Est-ce le début d’une nouvelle ère ? C’est ce qu’affirment en tout cas certaines personnes qui travaillent dans le domaine.
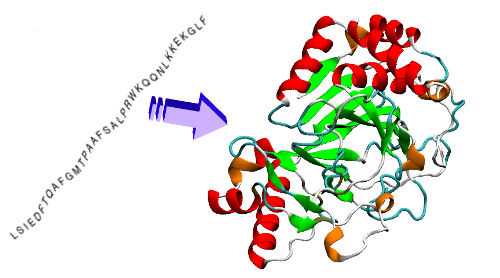
Pour comprendre à quel point tout cela est fascinant, il faut se rappeler que depuis que l’on a cartographié l’ensemble du génome humain il y a un peu moins de vingt ans, on connaît la séquence des gènes codant pour des protéines. Cela nous permet de connaître la suite d’acides aminés qui constitue ce que l’on appelle la structure primaire d’une protéine, c’est-à-dire le collier de perles d’acides aminés qui s’enchaînent l’un après l’autre pour la former. Mais ces acides aminés possèdent des charges électriques et des atomes qui s’attirent et se repoussent, de sorte que la chaîne d’acides aminées va très vite se replier sur elle-même pour former une sorte de gros globule en 3D aux milles fonctions toutes aussi essentielles pour la cellule les unes que les autres. Dans les neurones, les protéines forment par exemple tous les canaux ioniques qui rendent possible l’influx nerveux ou tous les récepteurs de neurotransmetteurs au niveau des synapses, rien que ça…
Depuis un demi-siècle on cherche à comprendre comment se replient les protéines pour acquérir leur forme tridimensionnelle qui va leur donner leur fonction dans l’organisme. C’est long et pas facile avec les approches expérimentales comme la cristallographie aux rayons X, la spectroscopie par résonance magnétique nucléaire ou la cryomicroscopie électronique. Et c’est carrément fastidieux avec des résultats très moyens avec les approches de modélisation informatique. Il y a juste trop de possibilités, trop de degrés de liberté. Prenons par exemple l’enzyme lysozyme qui est impliquée dans la défense contre les infections bactériennes et que l’on retrouve chez de nombreuses espèces animales. Chez la poule, il comporte 129 acides aminés. De combien de façons une suite de 129 acides aminés peut-elle exister ? Comme il y a 20 acides aminés de base à partir desquels sont formés les protéines, cela donne 20129, ou encore 10168 : 10 suivi par 168 zéros façons différentes de faire une protéine de 129 acides aminés ! Voyez-vous l’ampleur du problème ?
Rendu ici, j’aurais le choix entre plonger dans cette complexité et en avoir minimalement pour plusieurs pages à tenter de lui rendre justice. Ou bien dire que le temps me manque pour faire une telle recherche et vous laisser avec quelques généralités supplémentaires. Mais j’ai la chance cette semaine de pouvoir opter pour une solution intermédiaire qui allie un riche travail de vulgarisation scientifique et une économie de temps. Deux excellents rédacteurs de science pour un public curieux de ce genre de découverte vont en effet faire le boulot à ma place, et en français en plus de ça ! 😉
Car en lisant d’abord la traduction du magazine Pour la science de l’article de Ewen Callaway publié le 30 novembre dernier sur le site web du magazine Nature, je me suis dit que je ne pourrais jamais faire mieux que ça pour raconter cette histoire. Même impression en écoutant le vidéo Youtube de l’excellente chaîne Science Étonnante, animée par David Louapre, sur cette incursion spectaculaire d’AlphaFold dans le monde des protéines. Si vous avez 22 petites minutes et que vous voulez tout comprendre sur cette affaire, je vous suggère vraiment de visionner cette vidéo. Et si vous n’en avez pas encore assez, allez voir son billet de blogue sur le même sujet où il creuse davantage certains aspects que sa vidéo ne lui a pas permis de faire.
En terminant, comme l’a confié une personne qui travaille activement à déterminer des structures 3D de protéines de façon expérimentales (et non pas de les prédire avec un logiciel) à un de mes amis qui me l’a rapporté, il y aurait encore pas mal de pain sur la planche pour l’approche expérimentale. Car les protéines se rassemblent souvent en complexes de plusieurs sous-unités, ce qu’on appelle la structure quaternaire des protéines. Et dans ces complexes protéiques, les interactions avec d’autres protéines déforment leur structure et amènent un autre niveau de complexité qui rend leur analyse encore plus difficile. Bref, semble y avoir « encore de la marge », comme disait cette spécialiste.
Non classé | Comments Closed